
When a “loss” isn’t a loss: what a de novo GLUL start‑loss variant teaches us about variant interpretation and exome reanalysis
Despite increased knowledge, more than half of detected variants are classified as variants of uncertain significance (VUS), and in turn, more than half of patients undergoing exome or genome sequencing still receive a negative report. Thus, there is tremendous need to improve the interpretation of tricky variants. Start‑loss variants are often assumed to result in protein deletion and are frequently deprioritized during variant interpretation. Emerging evidence now challenges this assumption. A recently characterized de novo start‑loss variant in GLUL demonstrates how disrupted translational regulation can have surprising effects on protein function that drive autosomal dominant disease, and why unresolved exomes deserve systematic reanalysis as gene-disease knowledge evolves.
Presented during Illumina’s Grand Rounds webinar series, this case highlights two key lessons for variant scientists and laboratory geneticists:
- Start‑loss variants are not uniformly loss‑of‑function and may exert pathogenic effects through other mechanisms.
- Reanalysis is often the only path to diagnosis as interpretation frameworks incorporate new biological discoveries.
Why this case matters for variant interpretation
A dominant disease mechanism established only recently
Start‑loss variants are commonly interpreted as null. However, functional studies now show that certain GLUL start‑loss variants give rise to a truncated yet enzymatically active glutamine synthetase that escapes normal glutamine‑dependent degradation. This leads to pathologic glutamine accumulation and downstream neurotoxicity, an effect more consistent with toxic gain‑of‑function via protein stabilization than classic haploinsufficiency (Figure 1).
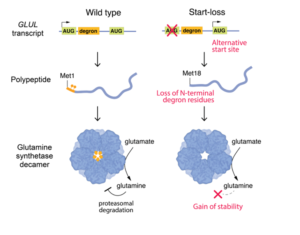
Figure 1 – Functional Consequences of a GLUL Start-Loss Variant
Until 2024, GLUL was not recognized as a cause of autosomal dominant neurodevelopmental disease. The first large series linking heterozygous de novo GLUL variants to Developmental and Epileptic Encephalopathy (DEE) was published only shortly before this case was reanalyzed, fundamentally altering how variants in this gene are triaged and interpreted.1
Clinical summary
Daniel is a five‑year‑old child with severe developmental delay, drug‑resistant epilepsy, and progressive white matter abnormalities. His neurological course includes early hypotonia, developmental regression, multiple seizure types (including myoclonus, absence, and focal seizures with apnea), and profound motor impairment. Brain MRI demonstrated diffuse cerebral atrophy and hypomyelination, and EEG showed disorganized background activity with focal epileptiform discharges.
The constellation of findings suggested a genetic etiology, initially raising suspicion for a metabolic or mitochondrial disorder.
Initial testing and the absence of a diagnosis
Standard genetic testing, including karyotype, chromosomal microarray, and clinical exome sequencing, was performed early in Daniel’s diagnostic journey and returned negative. For four years, no molecular explanation was identified.
Exome reanalysis reveals three variants
In 2024, Daniel’s parents requested reanalysis of his original exome data. Three heterozygous variants were identified:
- GLUL: start‑loss variant (pathogenic, de novo)
- HECW2: missense variant (variant of uncertain significance, inherited)
- HMGCL: stop‑gained variant (pathogenic, carrier state)
Variant interpretation: separating signal from noise
GLUL: the disease‑causing variant
The most compelling finding was a de novo start‑loss variant in GLUL, encoding glutamine synthetase, a critical enzyme in CNS ammonia detoxification and neurotransmitter cycling.
Pathogenicity was supported by multiple orthogonal lines of evidence:
- A disease mechanism now known to be protein‑stabilizing and dominant
- De novo occurrence confirmed by parental testing
- Functional data demonstrating altered regulation rather than loss of enzymatic activity
- Absence of the variant from population databases
- Strong phenotypic concordance with recently reported GLUL‑associated DEE cases
Downstream ATG codons can sometimes rescue translation in start‑loss variants, including the GLUL variants associated with DEE. However, functional data indicated this mechanism produced a truncated GLUL protein with pathological effects. The variant was therefore classified as pathogenic.
GLUL is an emerging autosomal dominant DEE gene whose disease mechanism was only recently established. Critically, this variant would have had an uncertain interpretation and may have never been prioritized before the GLUL disease mechanism was established.
HECW2: plausible but likely not causal
A heterozygous missense variant in HECW2, a gene associated with autosomal dominant neurodevelopmental disorders, initially appeared plausible based on phenotypic overlap. However, inheritance from an unaffected parent, lack of conservation at the altered residue, and absence from ClinVar reduced support for pathogenicity. The variant was classified as a VUS and not considered causative.
HMGCL: pathogenic but not explanatory
A pathogenic stop‑gained variant in HMGCL was also identified. However, HMGCL‑related disease is autosomal recessive and presents with severe neonatal metabolic decompensation. Daniel’s heterozygous status and clinical course were inconsistent with a causal role, and he was classified as an unaffected carrier.
Why the diagnosis was missed initially
Daniel’s case underscores a fundamental reality in clinical genomics: a negative exome reflects the limits of current knowledge, not the absence of a genetic cause.
The diagnostic breakthrough became possible only after peer‑reviewed publications in 2024–2025 established heterozygous GLUL variants as a cause of autosomal dominant DEE and clarified their molecular mechanism. These studies described multiple patients with highly concordant phenotypes and demonstrated transcriptional and regulatory disruption via RNA sequencing. 1,2,3
Broader implications for genomic practice
This case illustrates several field‑wide lessons relevant to variant interpretation and laboratory workflows:
- Start‑loss variants require mechanism‑aware interpretation, not automatic null assumptions.
- Newly established gene-disease relationships can retrospectively unlock diagnoses.
- Systematic exome reanalysis is essential in unresolved neurodevelopmental disorders.
- Family advocacy remains a powerful driver of diagnostic closure.
Because our knowledge of gene-disease relationships continues to expand, genomic data are not static, but rather, are a renewable diagnostic resource. As illustrated by Daniel’s case, reanalysis can transform uncertainty into clarity for families and advance the interpretive practice of the field.
References
-
Jones AG, Aquilino M, Tinker RJ, et al. Clustered de novo start‑loss variants in GLUL result in a developmental and epileptic encephalopathy via stabilization of glutamine synthetase. Am J Hum Genet. 2024;111(4):729‑741.
-
Carbonell E, Stenton SL, Ganesh VS, et al. Male proband with intractable seizures and a de novo start‑codon‑disrupting variant in GLUL. Hum Genet Genomics Adv. 2025;6(2).
-
Oh DE, Jang SS, Kim WJ, et al. Expanding the clinical and genetic spectrum of GLUL‑related developmental and epileptic encephalopathy. Sci Rep. 2025;15(1):35655.